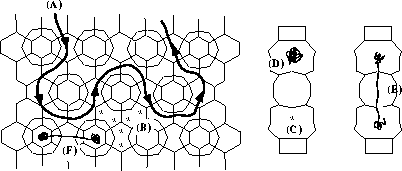
1. Molecular Dynamics of the Diffusion of Hydrocarbons in Zeolites.
We use an atomistic approach based on forcefields to simulate how hydrocarbons diffuse through the microporous structure of zeolites. I employ the general purpose DLPOLY code which I recommend for it is robust, portable, easily modifyable, and quick in serial and parallel machines. continued...
2. Quantum Chemistry Studies of Titanio- and Germanio-zeolites.
We study the new properties acquired by a silica framework when Ti and Ge atoms are introduced into it. Some of the new properties can be explained by the great distortion caused in the framework after the Ti/Ge replaces the original Si atom. This depends on how the structure can accommodate the strain, so different structures are expected to yield different properties -such as Lewis acidity- depending on its flexibility. We use standard HF and DFT methods under Gaussian95 by using basis sets of different qualities such as 3-21G**, 6-31G, 6-311-G, 6-31G**, among others, and clusters of medium size. Also we intend to explore the reactivity of these clusters in reactions involving charge transfer. continued...
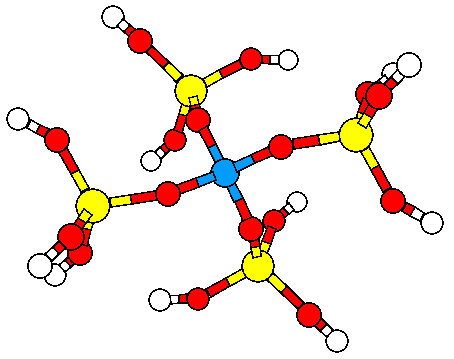
3. Study of electric fields in microporous materials.
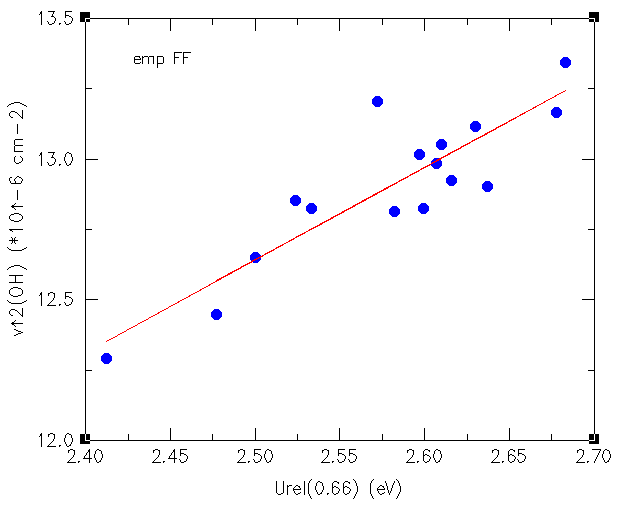
Electric charges are known to be responsible of nucleophilic and electrophilic attack and in general, chemical reactivity is explained in terms of electric charges. Organic molecules, when they react catalytically inside a microporous material are immersed in an electric field that can modify its chemical behaviour. In spite of this widely acknowledged principle, little has been reported on this topic and that is why we have conducted (in collaboration with Dr. Dewi Lewis) calculation on electric fields created by zeolites. We have been able to demonstrate how electric fields created by the microporous framework does influence the Brønsted acidity and have found a correlation with the square of the OH stretching frequency as shown in the figure below. continued...
4. Brønsted acidity of zeolites and SAPOs.
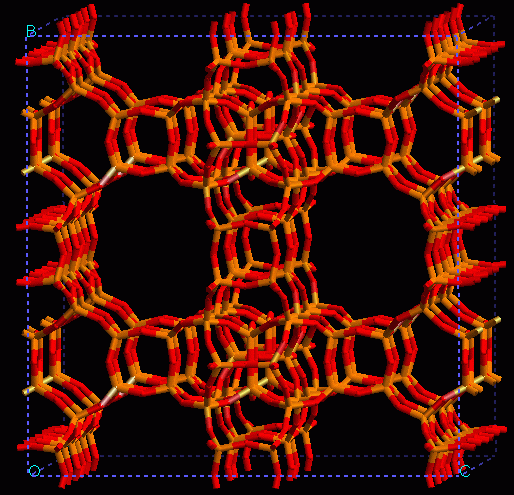
A long standing research project has been carried out by our group on this subject by using several computational techniques. A remarkable finding has been made on several zeolites and related microporous materials which seems to be of general application and it is the capability of atomistic forcefield methodologies to simulate OH stretching frequencies, that characterise Brønsted acidity. One of the examples considered has been the zeolite ITQ-7 shown below. continued...
5. Topological and geometrical characterisation of solids.
Zeolites and related microporous solids are three-dimensional four-connected nets where knots are tetrahedral atoms (Si,Al,P,Ge,...). A surprise comes when by only considering a few combination rules, more than 140 different materials have been characterised to this date, and hypotethical structures (that could be synthesised) are virtually countless. A part of the secret of this amazing variety comes from the flexibility of T-O-T angles, and so different topologies can -by deforming from equilibrium of dense structural binding shown by quartz and others- can be accommodated with reasonable strain that does not break the structure. continued...
6. Computer simulation of zeolite synthesis.
An interest does exist in synthesising new zeotype structures on the ground of -among others- potentially important industrial applications. Whether this topic has been traditionally regarded as a series of cooking recipes, the recent panorama is quite much changed and a general understanding is growing steadily. Even more recent is the rational use of computational tools in this respect and our approach comes from the atomistic forcefield approach that we have been using successfully to so many other research topics related to zeolites. continued...
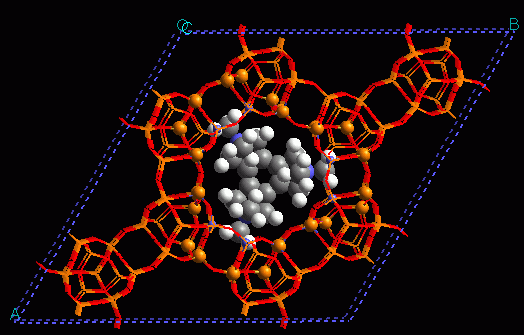
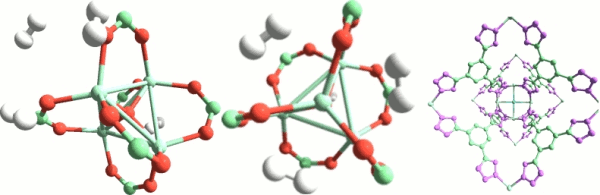
7. Metal-organic frameworks for adsorption and catalysis.
New microporous materials, metal-organic frameworks, have made their appearance in the chemistry scene very recently. Since their synthesis in Yaghi's group in 1999, the interest for these materials has experienced an exponential growth. A key aspect here is that bonds are covalent, hence strong and directional, and topologies are simple. Further, the reticular synthesis approach developed means that these materials can be designed on an aprioristic basis. In fact nowadays their number approaches one million. continued...